Welche Therapien gibt es?
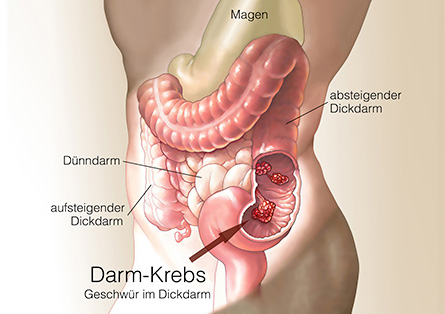
Bei einer nachgewiesenen familiären adenomatösen Polyposis besteht die einzige Möglichkeit, die Entstehung von Darmkrebs zu verhindern, in der vorsorglichen Entfernung des Dick- und Mastdarms. Wann diese prophylaktische Operation durchgeführt wird, ist individuell verschieden. Die Entscheidung darüber wird von den Untersuchungsbefunden, insbesondere von den Ergebnissen der regelmäßigen Darmspiegelungen, abhängig gemacht. Wenn es die Untersuchungsbefunde erlauben, wird empfohlen, die Pubertät abzuwarten.
FAP-Patienten sollten in Zentren für familiären Darmkrebs betreut werden, da sie auf die Betreuung, Diagnostik und Therapie von erblichem Darmkrebs spezialisiert sind. Informationen zu diesen Zentren finden Sie auf der Webseite der Deutschen Krebshilfe.
Im Wesentlichen gibt es drei Operationsmethoden der FAP:
Der ileo-anale Pouch
Der ileo-anale Pouch ist heute die international empfohlene Operationstechnik. Bei dieser Operation wird der Dickdarm vollständig entfernt. Der Schließmuskel bleibt allerdings erhalten. Aus dem freien Ende des Dünndarmes wird eine Tasche gebildet und an den Schließmuskel angenäht. Damit diese komplizierte Naht geschützt wird und in Ruhe ausheilen kann, wird vorübergehend ein künstlicher Darmausgang angelegt. Nach ein bis drei Monaten kann der künstliche Ausgang zurückverlegt werden. Nun übernimmt die Dünndarmtasche teilweise die Funktion des Dickdarmes und entzieht dem Stuhl Wasser. Allerdings kann sie diese Aufgabe nicht genauso perfekt erledigen wie ehemals der Dickdarm. Aus diesem Grunde haben Betroffene einen etwas erhöhten Flüssigkeitsverlust, der wieder ausgeglichen werden muss. Patienten, bei denen diese Operationstechnik angewandt wurde, haben durchschnittlich drei bis sechs Stuhlentleerungen pro Tag.
Die ileo-anale Anastomose
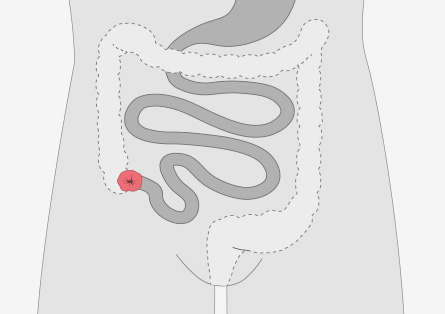
Auch bei dieser Operationstechnik wird der Dickdarm entfernt. Allerdings verbleibt der Enddarm und wird mit dem freien Ende des Dünndarmes verbunden. Diese Art der Operation kann nur durchgeführt werden, wenn der Enddarm polypenfrei ist. Dieser muss dann im Rahmen der Nachsorge engmaschig untersucht und sich eventuell bildende Polypen müssen entfernt werden, da diese bösartig entarten können.
Auch bei dieser Operation haben die Betroffenen häufigere Stuhlentleerungen.
Totale Proktokolektomie mit Ileostoma
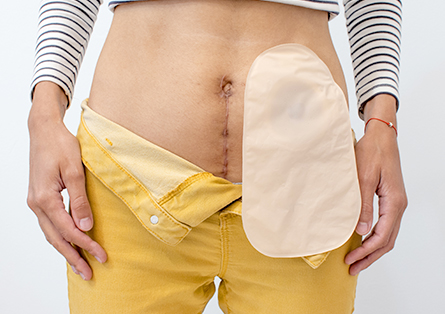
Bei diesem Eingriff werden der Dick- und Enddarm und der Schließmuskel vollständig entfernt und es wird ein dauerhafter künstlicher Darmausgang angelegt. Dieser Eingriff wird nur dann durchgeführt, wenn bereits ein bösartiger Tumor im unteren Enddarm vorliegt. Die beiden oben genannten Operationstechniken eigenen sich nicht, weil mit ihnen nicht sichergestellt werden kann, dass der Tumor vollständig entfernt wird.
Der künstliche Darmausgang, auch Stoma genannt, entsteht, indem das Dünndarmende mit einer operativ angelegten Öffnung in der Bauchdecke verbunden wird. Der Stuhl fließt dann nach außen ab und wird in einem speziell dafür entwickelten Beutelsystem aufgefangen. Viele Betroffene sind natürlich schockiert, wenn sie erfahren, dass sie für immer einen künstlichen Darmausgang erhalten. Doch die meisten kommen nach einer Eingewöhnungszeit in ihrem Alltag gut damit zurecht. Vor und vor allem auch nach der Operation stehen ihnen Spezialisten und Berater zur Seite. Sie schulen sie in der Stomapflege und helfen dabei, die geeignete Stomaversorgung zu finden. Heute gibt es moderne Hilfsmittel, die es den Betroffenen ermöglichen, ihren Alltag mit Job, Sport und Urlaub trotz Stoma zu meistern und auch wieder zu genießen.
Unter unserer Rubrik Stoma erhalten Sie noch ausführlichere Informationen zum Ileostoma.